美国FDA声明,开始启动对510 (k) 程序的改革
来源:天纵检测
|
作者:SKYLABS
|
发布时间: 2746天前
|
1919 次浏览
|
🔊 点击朗读正文
❚❚
▶
|
分享到:
天纵检测(SKYLABS)注意到:美国食品药品监督管理局(FDA)目前正在寻求对其510(k) 监管审查程序进行现代化改革。
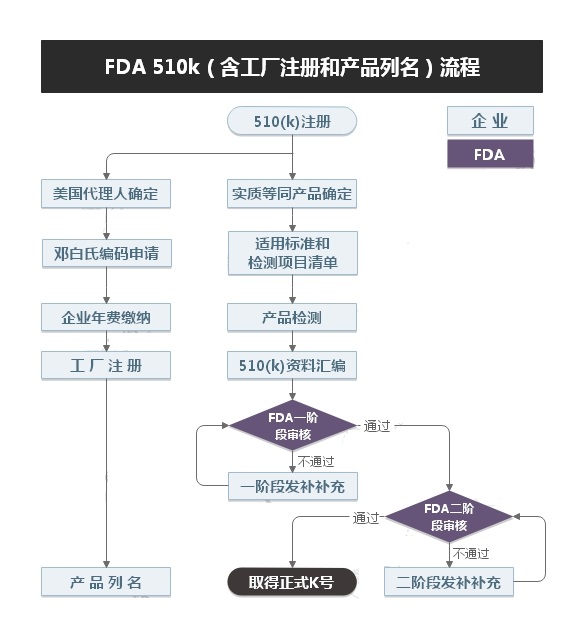
一个医疗器械进入美国市场商业销售,必须通过FDA的登记,且必须至少90天之前递交一个上市前通知,业内一般将其俗称“510(k)”程序。针对目前绝大部分一类和二类产品在完成企业注册、产品列名和GMP质量系统,完成510(k)申请后,即可获得FDA核准上市。
但近日天纵检测(SKYLABS)注意到:美国食品药品监督管理局(FDA)目前正在寻求对其510(k) 监管审查程序进行现代化改革。
FDA在2018年4月12日发布了一项关于扩展简化的510(k)程序:通过性能标准证明实质等同的草案,并计划于2019年初定稿;2019年财年投入使用安全网络(NEST);在接下来的几周内发布更新“de novo”程序的新法规。所有措施的目的是为了将改进后的510(k) 程序作为大多数产品的主要上市申请途径,以实现随着设备使用安全问题的出现,提高FDA的干预能力的目的,保障器械使用过程中的安全有效性。
对于40多年前的医疗器械修正法案实施前和实施后上市的许多医疗器械产品,在当时的技术水平与安全有效性考虑限制下,无法体现现在的先进技术,也不能更准确地反映当前人们对收益和风险的理解。为了应对新的特殊控制措施实施过程中的安全问题,需要将之前的某些产品升级为III级。
为配合510(k)程序的改进,FDA通过创新安全网,促进在监管决策中使用真实的证据,并引入新的从头监管措施。据悉由FDA和业界资助的安全网络,即国家卫生技术评估系统(NEST),将在2019财年投入使用。CDRH(设备仪器与放射健康中心,是FDA的下属机构)还打算在接下来的几周内发布一项新的法规来更新“de novo”程序。更新的目的是为了适应制造商对510(k)s的预期使用的增加,这是基于大部分的设备不断增长的批准请求。它们还设法通过编纂审查过程和确定要求中应包括的必要内容来提供一致性。
天纵君个人认为从FDA的相关声明来看,FDA的改革趋势似乎是对当前510(k)路径的简化版,它允许提交者主要依赖于符合共识标准的基线水平进行类比论证(而不是基于老旧的医疗设备)。对部分产品可进行简化的510(k)路径,但对于大多数设备类型继续使用标准510(k)申请,并保持对标准的更新,以确保设备技术保持现代化。




